
")
Microangiopathic hemolytic anemia (MAHA)
- Microangiopathic subgroup of hemolytic anemia caused by factors in small blood vessels (loss of red blood cells due to destruction).
- This occurs when red cells are forced to squeeze through abnormally narrowed small vessels.
Pathogenesis:
● A microvascular lesion that causes mechanical injury to circulate red cells.
● When cells quickly pass through the turbulent area of small blood vessels partially blocked by microthrombus or damaged endothelium, the RBC membrane is mechanically sheared.
● Upon shearing RBC membranes quickly reseal with the minimal escape of hemoglobin and schistocytes are formed.
● Thrombocytopenia occurs due to the consumption of platelets in the thrombi formed in the microvasculature
● Schistocytes in the peripheral blood film - Characteristic feature
● Grading of schistocytes
- Fragmented red cells as % of all red cells
- <1% - occasional
- 1%-3% - 1+
- 3%-6% - 2+
- 6%-12% - 3+
- > 12% - 4+
● Helmet cells, microspherocytes, polychromasia & nRBC
● Thrombocytopenia.

Causes of MAHA
• Thrombotic thrombocytopenic purpura(TTP)
• Hemolytic uremic syndrome(HUS)
• HELLP syndrome
• Disseminated intravascular coagulation
• Severe burns
• Malignant hypertension
• SLEI Scleroderma
• Drugs- Cyclosporin, Gemcitabine, Mitomycin —c
• C. difficile, Rickettsia rickettsii, B. anthracis.
Thrombotic Thrombocytopenic Purpura (TTP)
• Most common in adults, 4th decade, Female predominant
• Characterized by
1. MAHA
2. severe thrombocytopenia
3. Marked elevated serum LDH activity
• Neurological dysfunction, fever, and renal failure
• Deficiency of vWF cleaving protease known as a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13(ADAMTS- 13).
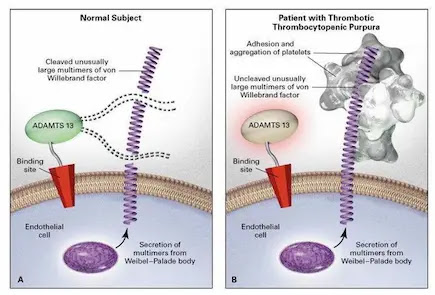
The proposed relationship between ADAM TS 13 lack of activity in vivo, excessive platelet adhesion and aggregation, and thrombotic thrombocytopenic purpura.

Pathogenesis of TTP
• ADAMTS13 serves an important antithrombotic function by preventing VWF excessive binding and activating platelets
• Platelets- VWF microthrombi block small blood vessels result in thrombocytopenia, ischemia in the brain, kidney, and other organs; and hemolytic anemia due to RBC rupture.
• Intravascular hemolysis and extensive tissue ischemia resulting in a striking increase in serum LDH.
Types of TTP
• Idiopathic
• Secondary
• Inherited
Idiopathic TTP
• No precipitating events
• Autoantibodies to ADAMTS13
• Autoantibodies are usually IgG class but can be IgM or IgA.
Secondary TTP
• Infection, pregnancy, surgery, trauma, inflammation, and diffuse malignant tumors may be caused by inhibiting the synthesis of ADAMTS13.
• Inhibitory reaction to ADAMTS13 occurs in conditions like hematopoietic stem cell transplantation; autoimmune disorders; HIV; drugs like quinine, ticlopidine, and trimethoprim.
Inherited TTP
• Upshaw-Schulman syndrome
• Severe ADAMTS13 deficiency by a mutation in the ADAMTS13 gene
• May present in infancy or childhood with recurrent episodes.
Laboratory findings of TTP
CBC:
- Decreased hemoglobin and platelets
- Increased reticulocyte count
Peripheral blood film:
- Schistocytes
- Polychromasia
- nRBC(severe cases)
Biochemical
- Increased LDH activity and serum total and indirect bilirubin
- Decreased serum haptoglobin level
- Hemoglobinemia
- Hemoglobinuria
- Proteinuria
- Hematuria
Treatment of TTP
• Idiopathic T TP- 80 to 90% of patients respond to plasma exchange therapy
• Corticosteroids are useful to suppress the autoimmune response
• Rituximab- in relapsing TTP
• Secondary TTP- do respond well to plasma exchange, not prognosis is poor except for TTP is related to autoimmune disease, pregnancy, and Ticlopidine use
• Inherited TTP- an infusion of fresh frozen plasma
Hemolytic uremic syndrome (HUS)
• It is characterized by MAHA, thrombocytopenia, and acute renal failure.
Pathogenesis
1) Endothelial injury and activation.
2) Platelet aggregation.
Both cause vascular obstruction and vasoconstriction => Precipitate distal ischemia.
Endothelial injury & activation
• Triggers can be :
– Bacterial endotoxins
– Cytotoxins
– Cytokines
– Viruses
– Drugs
– Antiendothelial antibodies
– Abnormal multimers or inhibitors of vWF
• Exfoliation of the endothelium exposes potentially thrombogenic subendothelial connective tissue.
• Decreased production of PgI2 and nitric oxide will enhance platelet aggregation and cause vasoconstriction.
• Activate endothelial cells, increase adhesion to white blood cells, and thrombosis.
• Endothelial cells produce vWF multimers, which are still abnormally large, and platelets aggregate.
Platelet aggregation
• With the congenital or acquired loss of ADAMTS- 13(a vWF cleaving metalloprotease) activity, very large vWF multimers persist in circulation and induce aggregation by activating platelet surface glycoproteins.
Types of HUS:
• Classic(Childhood) HUS
• Adult HUS
Classic HUS
• 75% in children after intestinal infection with E.coli that produce verocytotoxin.
• Verocytotoxin is similar to Shiga toxin.
• Most frequently associated with bloody diarrhea.
• Some traced to the ingestion of infected ground meat.
• One of the principal causes of acute renal failure in children.
Pathogenesis
• Related to Shiga-like toxin.
• Toxin causes:
– Increased adhesion of leukocytes.
– Increased endothelin production.
– Loss of endothelial nitric oxide.
– Lysis of endothelial cells (in the presence of cytokines (such as TNF)).
• Enhancement of both thrombosis and vasoconstriction- microangiopathy.
Verocytotoxin also binds to platelets and directly activates platelets.
Clinical features
• Sudden onset.
• Usually after a GI or influenza-like prodromal episode.
• Bleeding manifestations(hematemesis & melena).
• Severe oliguria.
• Hematuria.
• Microangiopathic hemolytic anemia.
• Prominent neurological changes in some patients.
Adult HUS
• In association with infection.
• In the antiphospholipid syndrome.
• As complications of pregnancy and contraceptives.
• Associated with vascular renal diseases.
• In patients receiving chemotherapy and immunosuppressive drugs.
• In typical (epidemic, classic, diarrhea-positive) HUS, the trigger for endothelial injury and activation is commonly Shiga toxin.
• In the genetic form of atypical HUS, the cause of endothelial damage appears to be excessive, inappropriate component activation.
Laboratory findings
• CBC
– Anemia
– Thrombocytopenia
– Peripheral smear checking for schistocytes,
burr cells, helmet cells, spherocytes and
segmented red blood cells
• LDH (elevated)
• Haptoglobin (decreased)
• Reticulocyte count (appropriate)
• PT/PTT (normal; differentiates from DIC)
• Stool tests
– Shiga toxin, E. coli O157: H7 test
• Urine Analysis
– Hematuria, casts
• LFT
– Increased indirect bilirubin
• Chemistry
– Creatinine, hyperkalemia (renal failure)
Disseminated intravascular coagulation (DIC)
• Defibrination syndrome or consumption coagulopathy
Generalized activation of hemostasis secondary to systemic disease
• Fibrin microthrombi partially blocks vessels and consumes platelets, coagulation factors, clotting control proteins, and fibrinolytic enzymes.
• Fibrin degradation products including D- dimer, become elevated.
• Acute and uncompensated DIC- deficiencies of multiple hemostasis components
• Chronic DIC- normal or even elevated clotting factors levels
• In chronic DICI liver coagulation factor production and bone marrow platelet production compensate for increased consumption
• Although DIC is a thrombotic process, the thrombus is small and ineffective, so systemic bleeding is the first or most obvious sign.
• Acute DIC is usually fatal and requires immediate medical intervention.
Causes
1. Sepsis and severe infection
- Bacterial infection (gram-negative sepsis, gram-positive infections, rickettsia)
- Toxins
- Viral (HIV, CMV, VZV, hepatitis virus)
- Fungal (Histoplasmosis)
- Parasitic (malaria
2. Malignancy
- Acute promyelocytic leukemia
- Acute myelomonocytic or monocytic leukemia
- Disseminated prostatic carcinoma
- Lung. Breast, stomach, pancreatic cancer.
3. Obstetric complications
- Amniotic fluid embolism
- Abruptio placentae
- HELLP syndrome
- Eclampsia
- Retained dead fetus syndrome
4. Massive tissue injury
- Severe/Major trauma
- Burns
- Extensive surgery
5. Systemic diseases
- ABO Transfusion Incompatibility
- Transplant rejections
- Malignant hypertension
- Severe Liver Disease
- Vasculitis
6. Post Cardiopulmonary Bypass
7. Heat stroke and hyperthermia

Pathogenesis of DIC
• Circulating thrombin is a major culprit activates platelets, activates
coagulation proteins and catalyzes fibrin formation
• The fibrinolytic system may be activated at the level of plasminogen.
• Endothelial cell damaged releasing coagulation active substances
• Leukocytes particularly monocytes may be induced to release tissue factor
• Fibrin monomers fails to polymerize and coats platelets and coagulation proteins creating an anticoagulant effect
• Plasmin circulates in plasma and digests all forms of fibrinogen and fibrin
• Fibrin degradation products labeled X,Y,I,D,I,E and D dimer are detectable in plasma exceeding 20,ooong/mL
• Symptoms of organ failure such as renal function impairment, ARDS, CNS manifestations
• May have skin, bone, and bone marrow necrosis
• Purpura fulminans is seen in meningococcemia, chicken pox and spirochete infections.
DIC profile
|
Test |
Reference interval |
Value in DIC |
|
Platelet count |
1.5-4-5 lac/μL |
<1.5 lac/μL |
|
Prothrombin time |
II-14sec |
> 14 sec |
|
PTT |
25-35 sec |
> 35 sec |
|
D-Dimer |
0-240 ng/mL |
> 240ng/mL, Usually 10,000 to 20,000 ng/mL |
|
Fibrinogen |
220-498 mg/dL |
<220 mg/dL |
Treatment of DIC
• Surgery, anti-inflammatory agents, antibiotics, obstetric procedures may normalize hemostasis I particularly in chronic DIC
• In acute DIC treatment falls into 2 categories
A) Therapy that slows the clotting process
B) Therapy that replaces missing platelets and coagulation factors
• UFH may be used for its antithrombotic properties but it may aggravate bleeding so careful observation and support needed
• Thawed frozen plasma provides all the coagulation factor and replaces blood volume lost
• Prothrombin complex concentrate, fibrinogen concentrate, and factor VIII concentrate may be used in place of plasma
• Repeated measurements of fibrinogen, PTI PTT
• Platelet transfusion if severe thrombocytopenia
• Red blood cells are administered to treat anemia
• In addition to proven systemic fibrinolysis, fibrinolytic therapy is contraindicated
HELLP syndrome
• Hemolysis, increased liver enzymes, and low platelet count.
• < 1% of all pregnancies but develops in approximately 10 % to 20% of pregnancies with severe preeclampsia( mc 3rd trimester)
• Exact pathogenesis is not known
• In pre-eclampsia, abnormalities in the development of placental vasculature result in poor perfusion and hypoxia
• Antiangiogenic proteins are released from the placenta that blocks the action of placental growth factors
• Continued vascular insufficiency leads to endothelial cell dysfunction causing platelet activation and fibrin deposition in the microvasculature, particularly in the liver.
• Anemia, biochemical evidence of hemolysis and schistocytes in peripheral blood film
• Platelet count is < 100x109 /L
• Increased Serum LDH activity
• Increased Serum aspartate aminotransferase activity
• Decreased platelet count, Increased LDH, Increased aspartate aminotransferase activity are major diagnostic criteria for the HELLP syndrome
• Therapy includes delivery of fetus and placenta as soon as possible, control seizures, hypertension, and fluid balance
• Mortality rate for mother — 3% to 5% and for the fetus - 9% to 24%.
 - Microangiopathic subgroup of hemolytic anemia caused by factors in small blood vessels (loss o…){kind=link}
Comments
Post a Comment